Research
Natural product synthesis

Natural products exhibit the most compelling combination of intriguing biological activity and sophisticated molecular architecture. These small molecules and peptidic compounds often promise to enhance our advance on therapies for debilitating or deadly diseases. The availability offered by nature, however, can often restrict initial biological activity studies, if not a probe of structure-activity relationships. Chemical synthesis, especially efficient chemical synthesis can hold the key to addressing the availability crisis in many cases.
Our program's efforts in new reaction development are always guided by the therapeutic potential of complex small molecules, and the application of new reactions to these targets are way-points in our studies. We have completed the total chemical synthesis of (+)-serratezomine A, hapalindoles K, A, G, (-)-verticilide, bassianolide, and feglymycin. These represent lycopodium, indole, depsipeptide, and peptide natural products. We have deposited samples of these natural products through the NIH CANVASS program. Each synthesis illustrates a strategy-level innovation supported by new tactical reactions.
Angew. Chem. Int. Ed. Engl. 2025, 64, e202508819
bioRxiv 2023 & Mol. Pharm. 2024, 105
ACS Med. Chem. Lett. 2021, 12, 1942
Proc. Natl. Acad. Sci. 2019, 116, 4810
Proc. Natl. Acad. Sci. 2016, 113, 14893
Angew. Chem. Int. Ed. 2011, 50, 7641
Catalysis
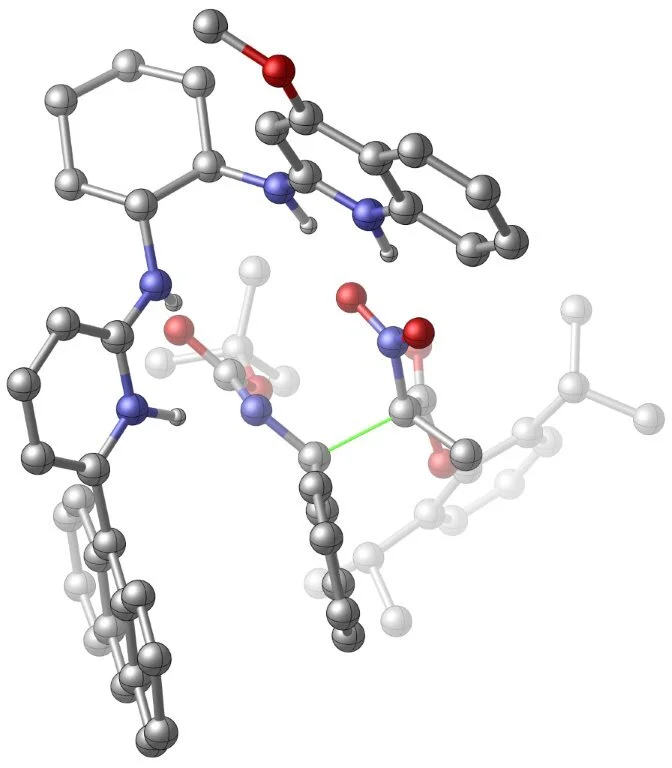
Enantioselective catalysis is essential to the concise preparation of functional groups at a chiral carbon with predictable control and high levels of selectivity. For many decades complex natural products, especially polyacetate and polypropionate classes, provided ample direction for the development of enantioselective oxidations and aldol reactions. Chiral metal-organic complexes were mainstays of catalyst designs, countless solutions were developed, and this technology was successfully applied to complex natural product synthesis. Similar target-derived inspirations have fueled our interest in chiral proton catalysis, but the opportunity to use small molecules in hydrogen bond catalysis has driven our development of a broad range of chiral amine and diamine syntheses using the aza-Henry reaction. Chiral proton catalysis, the name itself acknowledging the ligand-induced stereoselection, has grown to include a broad range of amidine-based reagents used as catalysts. The concept has been applied most extensively to azomethine (C=N) activation, but has evolved more recently to include alkene (C=C) activation. It is already clear that the large nature of proteins is not essential to achieve high levels of activation and enantiocontrol.
Our work has pioneered the specific use of polar ionic hydrogen bonds in catalysis. A mechanistic understanding of the factors affecting reactivity and stereocontrol in this type of catalysis has only recently developed, but contributes unique design principles to the broader topic of bifunctional catalyst design. A goal of highest priority is the discovery of general enantioselective reactions that defy the selectivity-generality paradox, and the development of a deeper understanding of the mechanism by which catalysts can be endowed with the broadest possible substrate scope.
J. Am. Chem. Soc. 2025, 147, 17584
J. Am. Chem. Soc. 2024, 146, 1269
Org. Lett. 2023, 25, 950
Chem. Eur. J. 2023, 29
Chem. Sci. 2022, 13, 7318
Chem. Sci. 2022, 13, 2614
J. Org. Chem. 2021, 86, 15606
ACS Catal. 2018, 8, 11926
Therapeutic development

Just as our program in reaction and reagent development is driven by structures discovered in nature by activity-based screening, the therapeutic development activities of companies and academic laboratories provide equally interesting small molecules with synthetically-confounding functional group arrays. These can exhibit additional challenges that result from drug development goals that include conformational restrictions (e.g. high levels of substitution at chiral carbon centers) or metabolic considerations (e.g. fluorine-carbon bonds). The availability of small molecules for testing biological hypotheses, and the adaptability of a synthesis to deliver important new candidates for lead development are drivers of route development to promising therapeutics. Our efforts have been focused on key challenges facing early stage therapeutic development in academic drug discovery. We have developed the first enantioselective routes to numerous small molecule therapeutics, including (+)-VNI, (-)-Nutlin-3, fluoro-lanicemine, LY411575, and a GlyT1 inhibitor. Each of these preparations readily produce gram (to decagram) quantities, and exhibit the convergency essential for use as a drug development platform. We establish collaborations across disease areas to use these materials in pursuit of answers to biological hypotheses.
ACS Med. Chem. Lett. 2025 16, 638
Chem. Sci. 2024, 15, 14977
J. Med. Chem. 2024, 67, 12205
Mol. Pharm. 2024, 105, 194
ACS Chem. Biol. 2023, 18, 2290
J. Mol. Cell. Cardiol. 2023, 180, 1
J. Pharm. Exptl. Ther. 2023, 385, 205
ACS Med. Chem. Lett. 2022, 13, 1755
Circulation 2022, 145, 1480
Circ. Res. 2021, 128, 321
Proc. Natl. Acad. Sci. 2019, 116, 4810
Umpolung amide synthesis (UmAS)

Amide synthesis is perhaps the most performed chemical reaction performed in laboratories worldwide. Its use is driven partly by the value and versatility of amides as constituent functional groups, as well as the importance of polyamide-containing compounds (e.g. peptides, proteins, antibodies). Remarkably, amide synthesis was long relegated to a single mechanistic paradigm based on nucleophilic amine reacting with electrophilic carbon in the key carbon-nitrogen bond-forming step. We discovered that alpha-bromo nitroalkanes and amines will react with the aid of electrophilic iodine (e.g. N-iodosuccinimide) and stoichiometric base (potassium carbonate) to form an amide. The hallmark of this reaction is polarity reversal, wherein the carbon is nucleophilic, and the nitrogen electrophilic, in the key carbon-nitrogen bond-forming step. The unique mechanism afforded us the opportunity to 1) mechanistically avoid alpha-carbon epimerization, and 2) create a new synthetic entry to chiral amides that leverages enantioselective catalysis against longstanding challenges in chiral amide synthesis. We have developed the most general platform to-date for aryl glycinamide synthesis. Additionally, we have developed an approach to D- or L-amino amide homologation of polyamides containing an aliphatic side chain. The reagent combinations deployed for UmAS have evolved from stoichiometric NIS, to 5 mol % NIS/O2, to recent work with KI/urea-hydrogen peroxide. One cocktail produces only inorganic salts as co-products, wastes that are readily washed away from the organic amide product.
Angew. Chem. Int. Ed. Engl. 2025, 64, e202508819
J. Am. Chem. Soc. 2024, 146, 1269
J. Am. Chem. Soc. 2022, 144, 16708
J. Org. Chem. 2022, 87, 5451
Cell: Chem 2019, 5, 1248-1264
Chem. Sci. 2019, 10, 1138-1143
Macrocyclooligomerization (MCO)
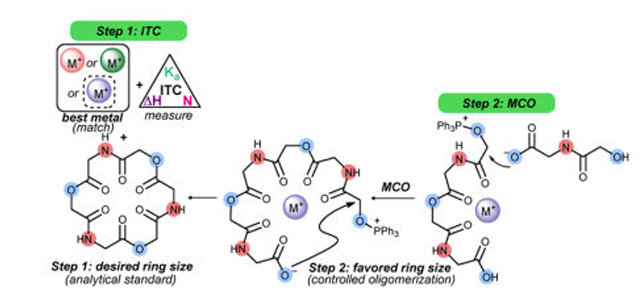
Macrocyclic structures are sought for their conformationally-limited presentation that can result in enhanced receptor binding. The functional complexity of macrocyclic small molecules often drive their activity, but the high step-counts required to prepare them ultimately limit their applications. Regardless, in order to probe the activity of complex small molecules, the ability to molecularly edit these structures and create investigational amounts ultimate determines the pace of dependent studies. We have turned our attention to oligomeric cyclic depsipeptides for their 1) representation among natural products, and 2) their modularity. As a class of functionally-diverse constrained macrocyclic compounds, we sought a method for their rapid preparation by sequential oligomerization/ macrocyclization (MCO). In late 2016, we reported the first successful Mitsunobu-based MCO to prepare cyclic depsipeptides with a range of ring sizes. Furthermore, we discovered that Lewis acidic salts could function as possible templates for MCO, causing perturbations in ring-size distributions. We now have examples where a single ring size can be enhanced to high yields (>85%), while other salts can favor a broad distribution of ring sizes (including a 60-membered ring cyclic depsipeptide) while providing useful investigational amounts of each. Finally, isothermal titration calorimetry measurements support the original hypothesis that the salt additives bind macrocyclic depsipeptides during formation: these salt-macrocycle measurements correlated with ring-size selectivity from the salt-modified MCO.
J. Org. Chem. 2021, 86, 7904
JACS 2018, 4560-4568
Proc Natl Acad Sci. 2016, 14893-7
JACS 2012, 15233-6